Qu'est-ce que l'alpha-galactosidase?
L'alpha-galactosidase est une enzyme qui appartient au groupe des hydrolases. Un autre nom de l'enzyme est la céramide trihexosidase.
L'enzyme se produit dans toutes les cellules humaines et clive la liaison alpha-D-glycosidique.
Une liaison alpha-glycosidique est une liaison entre un glucide tel que le galactose et un groupe alcool, par exemple.
Plus précisément, l'enzyme alpha-galactosidase se produit dans le lysosome des cellules et décompose les glycosphingolipides et les glycoprotéines en leurs composants individuels.
Si l'enzyme est défectueuse en raison d'une erreur génétique, le tableau clinique de la maladie de Fabry peut apparaître.

Tâche, fonction et effet de l'alpha-galactosidase
L'alpha-galactosidase est une enzyme et son travail est d'aider à décomposer les graisses spéciales. Ces graisses peuvent être absorbées dans le corps par la nourriture.
Ils se produisent dans pratiquement toutes les cellules du corps. La dégradation des graisses par l'alpha-galactosidase empêche la reproduction pathologique au sein des cellules. Le fonctionnement de l'enzyme correspond à celui d'une hydrolyse classique. Au cours de l'hydrolyse, la liaison chimique (liaison alpha-D-glycosidique) entre les composants individuels d'une molécule est divisée avec l'incorporation d'une molécule d'eau, ce qui donne deux produits.
Les propriétés typiques de l'enzyme accélèrent considérablement la réaction de division des graisses. En conséquence, plus de graisses peuvent être décomposées par unité de temps.
L'enzyme alpha-galactosidase émerge de cette réaction comme toutes les enzymes inchangées et dans la même quantité. La réaction catalysée de l'alpha-galactosidase a pour effet de réduire la quantité totale de glycosphingolipides dans l'organisme.
Les glycosphingolipides sont les noms des graisses qui sont décomposées par l'alpha-galactosidase.
Globalement, cela empêche la mort des cellules du corps en raison de l'accumulation de glycosphingolipides dans les cellules. Cela signifie que le tableau clinique de la maladie de Fabry ne se produit pas.
Où est-il fabriqué?
Les précurseurs de l'enzyme alpha-galactosidase sont synthétisés par les ribosomes des cellules. Ceux-ci libèrent une chaîne d'acides aminés, à partir de laquelle l'enzyme finale est créée plus tard, dans le réticulum endoplasmique.
Ici, la maturation a lieu jusqu'à l'enzyme finie. De là, les chaînes d'acides aminés sont transportées vers l'appareil de Golgi par des vésicules spéciales. De là, les enzymes peuvent être transportées par les vésicules dans les lysosomes ou à la surface cellulaire et libérées dans l'espace entre les cellules.
D'autres cellules peuvent désormais absorber les enzymes par l'intermédiaire d'un récepteur.
L'alpha-galactosidase a diminué
Une carence en enzyme alpha-galactosidase entraîne une dégradation réduite des graisses (glycosphingolipides) dans les cellules du corps.
Cela conduit à une accumulation de graisses dans les lysosomes des cellules.
Cette accumulation est mal tolérée par la plupart des cellules et a des effets néfastes sur elles. En conséquence, la mort cellulaire programmée se produit souvent. Cela peut se manifester par divers symptômes tels que des modifications de la peau et des vaisseaux sanguins ou des douleurs dans les mains et les pieds.
Maladie de Fabry
La maladie de Fabry est une maladie causée par une carence en alpha-galactosidase. La cause de la carence est généralement héréditaire et très rare.
La maladie n'est souvent remarquée que lorsque plusieurs symptômes apparaissent à la fin de l'enfance.
Il y a des changements cutanés ponctuels, des douleurs dans les mains et les pieds, un changement de sensation de température, une perte auditive, des changements dans les yeux et des protéines dans l'urine.
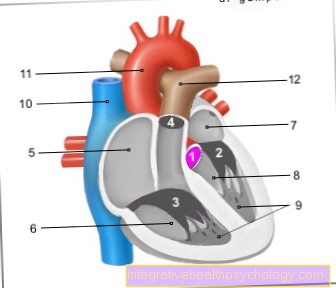
Les modifications des vaisseaux sanguins entraînent en particulier des menaces d'infarctus rénal ou d'insuffisance rénale dans les reins.
De plus, le cœur menace une crise cardiaque. Un accident vasculaire cérébral se produit également souvent. La thérapie consiste en la substitution précoce de l'enzyme par des comprimés. Une dialyse ou une transplantation rénale peut être nécessaire plus tard.
Pour des informations détaillées sur ce sujet, consultez: Maladie de Fabry
Pouvez-vous les remplacer par des comprimés?
S'il y a une carence en enzyme alpha-galactosidase, celle-ci peut être très bien contenue en prenant des comprimés, de sorte que pratiquement aucun changement ne soit perceptible.
Les comprimés contiennent des enzymes produites artificiellement qui fonctionnent exactement comme les propres enzymes du corps.
La substitution par des comprimés est la thérapie généralement utilisée en cas de déficit connu en alpha-galactosidase. Pour cette raison, la thérapie est également connue sous le nom de thérapie de remplacement enzymatique.
Parce que les cellules du corps ont des récepteurs de surface spéciaux qui reconnaissent l'enzyme, les cellules peuvent endocytoser l'enzyme dans leur plasma. Après absorption, les enzymes développent leur effet dans la cellule, ce qui réduit la quantité de glycosphingolipides.
Augmentation de l'alpha-galactosidase
Une quantité accrue d'alpha-galactosidase ne joue pas de rôle dans la médecine actuelle. Aucun effet négatif d'une grande quantité de l'enzyme alpha-galactosidase sur l'homme n'a été décrit.
Une quantité accrue d'alpha-galactosidase ne se produit généralement que si une quantité trop importante a été ingérée lorsqu'elle est remplacée par des comprimés.