Dystrophie myotonique
Synonymes
Dystrophie myotonique, maladie de Curschmann, maladie de Curschmann-Steinert
angl.: Dystrophie myotonique (musculaire).
introduction
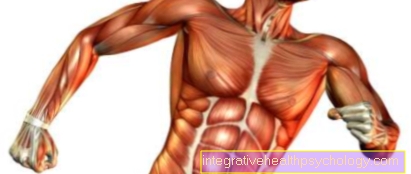
La dystrophie myotonique est l'une des dystrophies musculaires les plus courantes. Elle est associée à une faiblesse musculaire et à une fonte musculaire, en particulier dans la région du visage, du cou, des avant-bras et des mains ainsi que dans le bas des jambes et les pieds. La caractéristique ici est la combinaison des symptômes de faiblesse musculaire et de relâchement musculaire retardé après la contraction. Dans le même temps, des troubles d'autres systèmes corporels se produisent à des degrés divers: atrophie testiculaire et troubles hormonaux, arythmies cardiaques, opacification du cristallin (cataractes), troubles de la déglutition et de la parole, parfois les performances intellectuelles sont altérées au cours de la maladie. La dystrophie myotonique est une maladie héréditaire qui a tendance à apparaître plus tôt de génération en génération. Un changement caractéristique du matériel génétique sur le chromosome 19 s'est avéré être la cause de la maladie, ce qui conduit à une accumulation réduite d'une protéine qui assure l'intégrité de la membrane des fibres musculaires. Dans l'ensemble, la gravité de la maladie et l'implication de divers organes varient d'un patient à l'autre, et l'espérance de vie du patient est principalement déterminée par l'implication du cœur. Il n'y a pas de traitement causal de la maladie, les symptômes liés à la maladie des organes affectés peuvent être traités de manière symptomatique avec des médicaments. Il existe un risque accru d'anesthésie pendant les opérations, qui peut être réduit en utilisant certains médicaments.
définition
La myotonie dystrophique est la dystrophie musculaire la plus courante à l'âge adulte et est associée à une faiblesse musculaire, une atrophie et un relâchement musculaire retardé après l'activité. La maladie peut également affecter divers autres systèmes organiques. Cliniquement, il existe 3 formes différentes de dystrophie myotonique
- un congénital (inné) avec l'apparition des symptômes dans la petite enfance
- un adulte (forme la plus courante) à partir des deuxième et troisième décennies de la vie et
- une forme tardive qui commence à un âge avancé.
Épidémiologie
La fréquence de la dystrophie myotonique est donnée de 1/8000 à 1/20000. La maladie est héréditaire de manière autosomique dominante, ce qui signifie que les enfants des personnes touchées ont un risque de 50% de développer eux-mêmes la maladie, quel que soit leur sexe. Il y a une tendance à ce que la maladie apparaisse plus tôt de génération en génération et que la maladie soit plus prononcée ("Anticipation“).
cause première
La cause de la dystrophie myotonique est l'extension d'une section dans Chromosome 19 au-delà d'un certain niveau. Cela conduit à une production réduite d'une protéine qui est en partie responsable de la stabilité de la membrane des fibres musculaires. L'ampleur de l'allongement augmente avec l'hérédité de génération en génération et montre un certain lien avec l'apparition et la gravité des symptômes.
Symptômes
Dans la forme adulte de la dystrophie myotonique, combinaison d'une faiblesse musculaire progressive, en particulier des muscles de la main et de l'avant-bras, des pieds et du visage, avec une réaction de relaxation retardée des muscles après l'effort (myotonie). Cela peut également être observé en particulier dans les muscles de la main et des doigts, ainsi que dans les muscles du visage et de la gorge. B. Difficulté à relâcher un poing fermé ou à rouvrir les yeux fermés. Au fur et à mesure que la maladie progresse, elle peut entraîner des difficultés à avaler ou, en raison de l'atteinte des muscles respiratoires, une altération de la respiration.
Des arythmies sont observées dans le cœur, des palpitations et des trébuchements du cœur. Dans la région des organes génitaux, des symptômes tels que rétrécissement des testicules, menstruations absentes ou irrégulières et complications de la grossesse apparaissent.
De plus, les patients souffrent souvent d'opacités du cristallin (cataracte, «cataracte») et de surdité de l'oreille interne.
Dans la forme infantile de la dystrophie myotonique, les enfants atteints tombent tôt en raison d'une faiblesse musculaire ("bébé disquette«= Nouveau-né flasque), boire et s'épanouir et retarder le développement moteur. L'évolution de la maladie est généralement plus sévère. L'évolution tardive de la maladie peut, dans certaines circonstances, être atypique et, par exemple, être découverte uniquement dans le cadre de diagnostics complémentaires de cataracte ou dans le cadre d'une clarification familiale des cas de maladie chez la progéniture directe. Les patients atteints de dystrophie myotonique ont un risque accru d'anesthésie, car la maladie peut conduire à divers anesthésiques courants plus fréquemment à des complications, en particulier dans le domaine du système cardiovasculaire et de la respiration, que chez les patients en bonne santé. Il est donc important que l'anesthésiste soit informé de la présence de la maladie avant une opération.
Diagnostics différentiels
Les diagnostics différentiels incluent d'autres troubles myotoniques en fonction des symptômes dominants (relâchement retardé des muscles) ou d'autres dystrophies musculaires (fonte musculaire). De plus, les maladies du système nerveux peuvent entraîner une faiblesse et une perte des muscles contrôlés par les nerfs affectés.
Diagnostic
La présence de myotonie (relaxation retardée) des muscles avec atrophie musculaire (réduction de la taille et rétrécissement des muscles) est cliniquement révolutionnaire. Lors de la mesure de l'activité musculaire électrique dans l'EMG (électromyogramme), une constatation caractéristique du faible niveau des éruptions cutanées individuelles (comme dans les dystrophies musculaires) et l'apparition de séries de décharge rapide (comme dans la myotonie, soi-disant "Bruit de bombardier en piqué).
Le test sanguin montre les symptômes de la mort des cellules musculaires (augmentation de la valeur d'une enzyme musculaire, «CK») et éventuellement les conséquences d'une atteinte du système endocrinien (diminution des valeurs des hormones sexuelles). L'examen génétique humain permet de détecter l'élongation d'une section du chromosome 19 dans les cellules sanguines, caractéristique de la dystrophie myotonique. Si le diagnostic de dystrophie myotonique a été posé, il est important de clarifier la fonction du cœur au moins au moyen d'un électrocardiogramme (électrocardiogramme) afin de trouver des indications d'un éventuel trouble fonctionnel à un stade précoce.
thérapie
Un traitement causal de la dystrophie myotonique n'est actuellement pas possible. Les arythmies cardiaques peuvent être traitées avec des médicaments et, si nécessaire, avec un stimulateur cardiaque, et les spasmes musculaires avec des médicaments qui ont un effet stabilisateur sur l'état d'activation de la cellule musculaire. S'il existe des troubles hormonaux, ceux-ci peuvent également être traités avec des médicaments. Dans une certaine mesure, la thérapie physique peut être utilisée pour contrer les symptômes de faiblesse musculaire afin d'assurer la mobilité du patient et d'éviter une mauvaise posture.
prévoir
La maladie progresse lentement et varie d'un patient à l'autre. L'espérance de vie des patients est réduite à une moyenne de 50 à 60 ans et la mort survient souvent Insuffisance cardiaque une.
















.jpg)