Syndrome d'Ehlers-Danlos
Synonymes

EDS, syndrome d'Ehlers-Danlos-Meekeren, syndrome de Van Meekeren, Fibrodysplasia elastica generalisata, dermatolyse, cutis hyperelastica, "peau de caoutchouc", etc.
Français: Laxité articulaire congénitale multiple
Engl.: Syndrome de Danlos, syndrome de Meekeren-Ehlers-Danlos, syndrome de Tchernogubov, syndrome de Sack, syndrome de Sack-Barabas, syndrome de Van Meekeren I.
Russe: syndrome de Tchernogubov
Définition / introduction
le Syndrome d'Ehlers-Danlos (EDS) résume un groupe de hétérogènes, génétiques Maladies du tissu conjonctif ensemble, causés par des perturbations dans la synthèse du collagène, une protéine structurelle du Tissu conjonctif, symptômes conditionnels et caractéristiques du peau, Les articulations et les organes internes exposition.
la fréquence
le Syndrome d'Ehlers-Danlos c'est rare. La prévalence dans la population totale est de 1: 5000; 90% d'entre eux en font partie Types I, II et III touchés (30% chacun), et environ 10% de type IV. Les autres formes sont rarement observées.
le Types I-III devenir dominance autosomique hérité, c'est-à-dire il faut juste un gène défectueux pour que la maladie éclate. Les autres gars vont autosomique récessifc'est à dire. il doit y avoir deux gènes défectueux, ou Lié à l'Xc'est à dire. Transmission des chromosomes sexuels, héréditaire.
l'histoire
cela a été décrit pour la première fois Syndrome d'Ehlers-Danlos dans l'année 1668 de Job Janszoon de Meekeren (1611-1666), chirurgien d'Amsterdam. Il avait découvert le symptôme d'une extensibilité anormale chez un Espagnol qui pouvait tirer la peau de son menton jusqu'à ses yeux et sur sa poitrine. Cependant, il n'a observé aucune autre anomalie.
Première 1891 créé le dermatologue Tchernogubov une description complète du tableau clinique, y compris l'atteinte articulaire et vasculaire, c'est pourquoi dans la médecine russe
Littérature technique à ce jour le nom «Syndrome de Tchernogubov» est commun.
D'autres descriptions ont suivi 1901 par le dermatologue danois Eduard Ehlers (1863-1937) et 1908 par le dermatologue parisien Henri A. Danlos (1844-1912). Ce n'est qu'en 1933 que "Syndrome d'Ehlers-Danlos" comme le nom de la maladie prévaut.
1949 il y a eu des premiers aperçus sur la fréquence familiale de la maladie et 1972 était une erreur génétique liée à cela Syndrome d'Ehlers-Danlos découvert. En 1986, une classification préliminaire en 10 types a été établie, qui a été modifiée en 1997 en une version simplifiée avec la subdivision en six types principaux.
causes
La cause de la maladie est un Défaut génétique. Il y a un changement (mutation) dans les gènes qui composent la protéine structurelle Collagène décrire sur le génome humain, l'ADN. La mutation conduit à une structure modifiée et / ou à une synthèse réduite du collagène, ce qui conduit à une résistance réduite de l'ensemble du tissu conjonctif.
Tous les deux Types I et II c'est une mutation du gène collagène V, dans laquelle Types IV une mutation dans Collagène III.
Symptômes
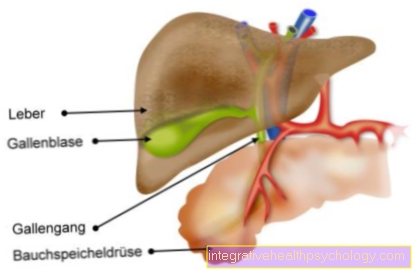
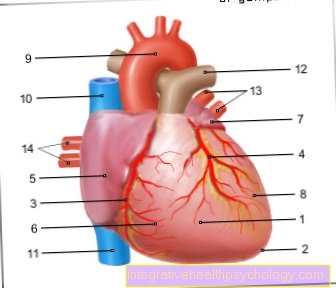
En raison de la synthèse de collagène perturbée et réduite, le Syndrome d'Ehlers-Danlos les parties du corps particulièrement riches en tissu conjonctif: les peau, Les articulations et Vaisseaux sanguins. Le tissu conjonctif manquant de force, il est trop extensible et se déchire très rapidement, ce qui est particulièrement vrai pour les vaisseaux sanguins trop petits, mais parfois trop saignement massif peut mener. Une complication importante est la formation de renflements dans les vaisseaux, ce que l'on appelle. Anévrismes, avec risque de rupture.
le Symptôme principal de la peau est le plus prononcé Cutis hyperélastiquesdu côté de la cou, plus de Les articulations et aussi dans visage peut être soulevé jusqu'à 4 cm ou plus. Après avoir lâché prise, il revient immédiatement à sa position de départ, c'est pourquoi il porte le nom "Peau en caoutchouc" portant. En général, la peau est visiblement fine (comme le papier à cigarette), douce et veloutée ("Peau de guimauve").
Les plaies montrent une cicatrisation retardée, de sorte que les sutures mettent 3 à 4 fois plus de temps à guérir. Atrophiques ou hypertrophiques, les inférieurs se développent souvent à partir des coutures cicatrice. En outre, il y a formation de renflements cutanés remplis de liquide (succulents) (pseudotumeurs mollusoïdes) sur les zones fortement sollicitées du corps, telles que Articulation du genou et du coude, à la formation de coussinets de cheville ("Coussinets d'articulation") sur Dos de la main et du pied et de nodules sur le talon.
le Les joints peuvent être trop étirés (Hyperflexibilité), souvent déplaçables dans des directions non désirées et manquant de force en raison de ligaments articulaires desserrés (Laxité ligamentaire). Cela peut provoquer des mouvements inhabituels, tels que ceux auxquels on pourrait s'attendre "Contorsionnistes" sait. Les articulations ont tendance à Contorsions (Luxations) et les malpositions. Ceux sont particulièrement touchés épaule- et Articulations de la cheville, les Rotule (rotule), les Articulation temporo-mandibulaire (Articulation temporo-mandibulaire) et plus rarement que Articulation du coude. La documentation de la surmobilité des articulations (hypermobilité) est réalisée par le Le score de Beightonqui confirme l'hypermobilité sur 5 des 9 points possibles.
D'autres symptômes des articulations sont des problèmes articulaires généralisés, douleur chronique au cou, bouge toi- et Douleur à la hanche, Joint et Douleurs musculairesqui sont difficiles à traiter. Parfois des points douloureux ("Ponits tendres"), qui sont définies comme une zone qui réagit douloureusement à des charges de pression de 4 kg ou moins. De plus, il existe un risque accru de fractures en raison d'une réduction de la masse osseuse associée à une structure osseuse anormale.
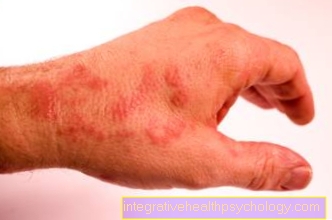
En raison de la fragilité du tissu conjonctif des vaisseaux sanguins, il existe une tendance prononcée à Hématomes, spontanément ou à la suite d'un traumatisme,
principalement dans les zones à risque de blessure. Ceci est suivi d'un typique dans les zones touchées pigmentation brune.
Après des blessures, on observe une tendance à saignement prolongé avec des valeurs de coagulation normales. La fragilité des gros vaisseaux sanguins peut être déclenchée par l'effort, les accidents, grossesse ou naissance conduisent à des saignements massifs et potentiellement mortels.
Étant donné que les autres structures du tissu conjonctif sont également inférieures, elles peuvent devenir trop Viscères (Hernie / hernie inguinale)), courbure de la colonne vertébrale (La scoliose), Des fissures (rupture) du Intestins et le utérus (Utérus), sacs (Anévrisme) des vaisseaux sanguins et la constriction des poumons due à l'air libre dans la poitrine (Pneumothorax) viens.
Dans de rares cas, des changements oculaires sont associés à l'EDS tels que Astigmatisme (astigmatisme) ou étoile verte (glaucome) observer.
diagnostic
Le diagnostic est posé sur la base de l'aspect clinique, des symptômes et est complété par un examen familial (antécédents familiaux). De plus Biopsie cutanée dans lequel le tissu cutané retiré est examiné à l'aide d'un microscope électronique et sa structure de collagène est évaluée. La différenciation entre les différents types de Syndrome d'Ehlers-Danlos a lieu au moyen d'une analyse de séquence de l'ADN.
Classification / types
Type I, II: Mec classique; Héritage: autosomique dominant; Principaux symptômes: Hyperélasticité et fragilité de la peau, cicatrices atrophiques, hypermobilité articulaire; Cause: trouble de la formation du collagène V
Type III: Type hypermobile; Héritage: autosomique dominant; Principaux symptômes: hypermobilité articulaire généralisée, atteinte cutanée (hyperélasticité et / ou peau douce et vulnérable); Cause: trouble de la formation du collagène V
En savoir plus sur ce type important sur: Syndrome d'Ehlers-Danlos type III
Type IV: Type vasculaire; Héritage: autosomique dominant; Principaux symptômes: peau fine et translucide, ruptures des artères, des intestins et de l'utérus, tendance prononcée à l'hématome; Cause: trouble de la formation du collagène III
Type V: correspond au type I.
Type VI: Type cyphoscoliotique; Héritage: autosomique récessif; Principaux symptômes: diminution de la tension du Musculature déjà à la naissance ("Bébé disquette"), retard de développement des réflexes de maintien et de soutien, flexion latérale du Colonne vertébrale (Scoliose), Cause première: Déficit en Lsysl hydroxylase
Type VII A / B: Type arthrochalastique; Héritage: autosomique dominant; Principaux symptômes: hypermobilité généralisée sévère des articulations avec luxations répétées, luxation congénitale bilatérale de la hanche; Cause première: Trouble du collagène de type I
Type VII C: Type dermatosparactique; Héritage: autosomique dominant; Principaux symptômes: fragilité cutanée prononcée, relâchement cutané, Cause première: Déficit en procollagène I peptidase N-terminal
Thérapie et prophylaxie
Aucun causal toujours un thérapie symptomatique est actuellement possible, de sorte que la prophylaxie des dommages consécutifs est au premier plan. Les blessures et un stress accru sur les articulations doivent être évités. Alors devrait être certain des sportsqui sont associés à un risque accru de blessures ne sont pas exercés. En raison du risque accru de complications grossesse et naissance tous les deux Types I, II, IV et VI une surveillance étroite est nécessaire.
Il doit également être utilisé avec le rhume traitement antitussif et généralement au Régulation de la consistance des selles être respecté, car une telle chose Rupture du côlon (Rupture du côlon) et un Pneumothorax peut être évité. Grâce à une physiothérapie précoce, en particulier chez les enfants, les articulations trop étirées peuvent être stabilisées, ce qui conduit à l'atténuation des symptômes de l'ensemble du système musculo-squelettique.
Blessures des précautions particulières doivent être prises et les opérations ne doivent être effectuées qu'en cas d'urgence car la cicatrisation des plaies prend 3 à 4 fois plus de temps que d'habitude.
prévoir
Patient avec Syndrome d'Ehlers-Danlos ont généralement une espérance de vie normale. Cependant, la maladie est progressive, donc elle conduit à une détérioration toujours croissante de la santé. Les plaies cutanées et les luxations articulaires affectent la qualité de vie du patient, tandis que la rupture des gros vaisseaux peut mettre la vie en danger.
Espérance de vie
Le syndrome d'Ehlers-Danlos est une maladie chronique pour laquelle il n'existe toujours pas de traitement causal et donc pas de cure. Cela signifie que, compte tenu de l'état actuel de la technologie médicale, il n'y a aucun moyen de faire quoi que ce soit sur les causes du syndrome d'Ehlers-Danlos et de le guérir complètement. Malheureusement, on n'est toujours pas en mesure de combattre et de traiter les symptômes qui surviennent. Le patient concerné ne peut qu'être encouragé à toujours faire attention dans la vie quotidienne à ne pas trop solliciter les articulations et à éviter si possible les blessures cutanées. Les interventions chirurgicales ne doivent être effectuées qu'en cas d'urgence et en l'absence d'alternatives adéquates.
Dans la plupart des cas, il est progressif avec une aggravation croissante des symptômes et des déficiences dans la vie quotidienne du patient. Selon le type de maladie, la maladie a des effets différents sur la vie des personnes touchées. Les modifications des articulations entraînent parfois de l'arthrose et de l'arthrite dans la petite enfance, de sorte que les enfants apprennent plus tard à marcher et que leurs pieds peuvent être déformés. Avec le risque accru de décollement de la rétine ou d'hémorragie rétinienne, la vue est également compromise.
La force des symptômes finalement prononcés chez chaque individu dépend fortement du type de syndrome d'Ehlers-Danlos et ils peuvent également varier considérablement au sein des sous-types individuels. Pour la plupart des types de syndrome d'Ehlers-Danlos, l'espérance de vie est normale. Chez les patients atteints du syndrome d'Ehlers-Danlos de type IV, qui affecte les vaisseaux, l'espérance de vie est considérablement réduite en raison de complications graves, telles que le risque de rupture spontanée d'une artère, en particulier de l'artère principale (déchirure aortique moyenne) ou du gros intestin.
Il est d'environ 37 ans pour les femmes et 34 ans pour les hommes.
Une espérance de vie réduite peut également être supposée avec le syndrome d'Ehlers-Danlos de type VI.